

Todo el mundo ha oído hablar del físico Stephen Hawking y sus importantes contribuciones al conocimiento de los agujeros negros en el espacio. Pero no muchos saben que en su vida tuvo que enfrentarse también a otro tipo de agujero negro más mundano, como podríamos denominar al “pozo sin fondo” que supone sufrir una enfermedad neurodegenerativa como la que él padecía: la esclerosis lateral amiotrófica (ELA).
Esa misma enfermedad es la que tomó la vida de Paola Roldán el pasado lunes, la ecuatoriana que logró que la eutanasia se despenalizara en el país tras una intensa lucha que llevó adelante combatiendo el dolor y la inmovilidad. Este reportaje publicado en The Conversation ahonda en las profundidades de esta enfermedad tan compleja.
ALGO DE HISTORIA SOBRE LA ELA
El nombre ELA proviene de las palabras “esclerosis”, que significa endurecimiento o cicatrización de un tejido y “lateral”, en referencia a la región afectada de la médula espinal, ocupada por las neuronas motoras de las que parten los “cables” o axones que llevan el impulso nervioso hasta las fibras musculares y que son las responsables de los movimientos voluntarios del organismo. Por último, “amiotrófica” proviene del griego “a” (negación) y de los términos “mio” y “trófica”, es decir, falta de nutrición en el músculo, en referencia a que éste no recibe señales nerviosas y se va degenerando poco a poco hasta perder su función.
La ELA fue identificada en 1869 por el neurólogo francés Jean-Martin Charcot, y ya en 1874 empezó a usarse el nombre con el que la conocemos hoy día. Sin embargo, fue en 1941 cuando la ELA empezó a ser conocida internacionalmente con el nombre de enfermedad de Lou Gehrig, por el nombre de un famoso jugador de béisbol del equipo de los míticos Yankees de Nueva York que la padeció, apodado “caballo de hierro”.
SÍNTOMAS DE LA ENFERMEDAD
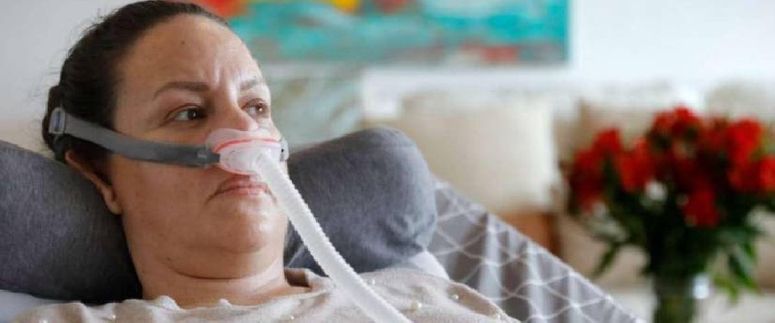
La ELA es una enfermedad neurodegenerativa que suele aparecer más frecuentemente a partir de los 50 años. Comienza con debilidad en manos y pies así como dificultad para caminar y realizar las actividades de la vida diaria, siendo habituales los tropiezos y caídas. Más tarde aparecen otros síntomas como dificultad en el habla y al tragar, además de calambres musculares y espasmos en brazos, hombros y lengua.
Por si fuera poco, más de un 5% de los pacientes de ELA suele sufrir también demencia frontotemporal, al verse afectadas neuronas del cerebro relacionadas con el comportamiento y la personalidad.
Finalmente alcanza a los músculos del diafragma y se acaba deteriorando la función respiratoria, lo que suele ser la causa del fatal desenlace de la enfermedad a los pocos años de aparecer los primeros síntomas.
 20.52.32.png)
NO HAY UNA SOLA ELA SINO MÚLTIPLES VARIANTES DE ELA
Conocer a nivel molecular una enfermedad suele ser la base para el posterior descubrimiento o desarrollo de medicamentos frente a ella. Y una vez conocidas las bases moleculares, se necesita mucha investigación para seguir avanzando.
El problema en el caso de la ELA es que son muchos los procesos celulares afectados que se han descrito en las células de los pacientes. Entre ellos, el procesamiento de moléculas de ARN, el tráfico de proteínas en el interior de la célula, el plegamiento y recambio de proteínas o la regulación de la muerte celular programada.
En total hay más de 40 genes identificados cuya mutación podría llevar a la aparición de ELA. Pero cada paciente suele tener mutado uno solo de estos genes.
Esto quiere decir que no hay una ELA, sino múltiples variantes de ELA, cada una con sus propias particularidades a nivel molecular, según los genes implicados.
CUANDO LA ELA SE HEREDA
A pesar de ello, sólo en el 5-10 % de los casos ha podido confirmarse que la ELA es heredada. Hablamos entonces de ELA familiar, y en estos casos la enfermedad está causada por mutaciones de los genes SOD1 y C9orf72. La herencia suele ser autosómica dominante, lo que significa que con sólo heredar una copia del gen mutado de nuestra madre o nuestro padre podríamos presentar la enfermedad.
Sin embargo, en muchos casos podemos tener un gen mutado sin que aparezca la enfermedad, por lo que podemos decir que la ELA es a veces una enfermedad con penetrancia genética limitada.
El gen SOD1 da lugar a una proteína que destruye radicales libres que son tóxicos y se producen de manera habitual por el metabolismo celular. En determinadas circunstancias, esta proteína se acumula formando agregados que impiden que ejerza su función, algo que se ve agravado cuando existen cambios en la secuencia del gen. Algunos estudios han mostrado a grupos de militares con una mayor probabilidad de desarrollar ELA, lo que podría ser debido a la exposición a tóxicos.
Por otro lado, el gen C9orf72 hace mención de que es el gen número 72 del cromosoma 9 humano. Esta falta de nombre propio ya da a entender el desconocimiento parcial que hay sobre su función. Sí que se sabe que el problema principal suele estar en la repetición descontrolada de parte del gen, como ocurre con otras enfermedades neuronales como el Huntington o la ataxia de Friedreich.
Solo en el caso de SOD1 se conocen más de 100 mutaciones distintas, cada una de las cuales puede estar asociada con un tipo distinto de ELA. Algunas se relacionan con la aparición de síntomas más tempranos, otras con una esperanza mayor de vida, e incluso con una mayor propensidad a padecer la enfermedad, sin que la probabilidad llegue a alcanzar el 100 %.
En el caso de la ELA familiar –en torno al 90 % de los casos–, la cantidad de genes y mutaciones implicadas complica aún más el conocimiento de su afectación molecular y progresión.
 21.09.24.png)
COMPARTE SÍNTOMAS CON OTRAS ENFERMEDADES NEURODEGENERATIVAS
La ELA es una de las enfermedades neurodegenerativas más frecuentes que se conocen. Aparece 1,75 veces por cada 100.000 habitantes en la población mundial, con 3 nuevos casos al día en países como España. Pero la sintomatología descrita en la ELA la sufren diariamente pacientes de otras múltiples enfermedades que implican degeneración neuronal, como la atrofia muscular espinal, la esclerosis múltiple, la enfermedad de Huntington o el párkinson, entre otras muchas, las cuales afectan a miles de personas.
Estas otras enfermedades no sólo comparten síntomas, sino a veces también genes implicados en su sintomatología. Y todo esto hace que a veces hasta se compartan posibles tratamientos.
GASTOS A ENFRENTAR E IGUALDAD REAL
La rápida progresión de estas enfermedades hace que los afectados y sus familias tengan que afrontar gastos extraordinarios para mantener una mínima calidad de vida. A las imprescindibles ayudas de ortopedia que incluyen sillas de ruedas, grúas de traslado o ayudas para el aseo, se añaden las ayudas para la alimentación y el mantenimiento de las funciones respiratorias, así como de asistencia personal. Y todo ello sin contar con los desplazamientos, que ya de por si son complicados si no se dispone de vehículo adaptado, lo que impide en muchas ocasiones el ocio o el acceso al trabajo.
Sthephen Hawking pudo salvar ese agujero negro durante los 55 años en que convivió con la ELA, algo muy inusual en esta enfermedad, gracias a su gran capacidad adquisitiva. Sin embargo, los pacientes de enfermedades neurodegenerativas deben hacer frente a los gastos de la enfermedad mayoritariamente con sus propios y más limitados recursos.
En Estados Unidos fue un jugador de béisbol quien puso el foco en la ELA, lo que hizo que se conociera la enfermedad y que se empezaran a poner recursos para su tratamiento. En España, un jugador de fútbol, Juan Carlos Unzué, está consiguiendo el mismo efecto. Ojalá sirva para que más pronto que tarde se empiece ayudar a conseguir la igualdad real de oportunidades para los pacientes de enfermedades neurodegenerativas.
(*) Profesor Titular de Universidad e Investigador en Bioinformática, Universidad Pablo de Olavide. Este artículo se publicó originalmente en: https://theconversation.com/la-esclerosis-lateral-amiotrofica-es-como-un-agujero-negro-225494.
Last modified on 2024-03-13